New X-ray Based Approaches to Probing Protein Structure and Function
The revolutions in molecular biology and molecular medicine are driven in part by the availability of high-resolution structures of proteins and other biological macromolecules. These structures provide insight into molecular function and a basis for rational approaches to the design of new medicines. However, nearly all such structures are determined at cryogenic temperatures, using either X-ray cryocrystallography or cryoelectron microscopy. Proteins under biological conditions exist in an ensemble of conformations that reflect their complex and multi-tiered energy landscapes, yet the overwhelming majority of X-ray structures are refined to a single conformation. We have been developing experimental methods that allow X-ray study of protein conformational ensembles in the presence of liquid water at temperatures down to 200 K, and for analyzing and interpreting this data. Small-angle X-ray scattering (SAXS) provides low resolution but high precision information about the structure of and interactions between biomolecules and their binding partners. We made the first successful demonstration of SAXS using cryocooled biomolecules, and are now working to develop cryoSAXS into a general purpose method that dramatically reduces sample volumes and data collection time per measurement and facilitates high-throughput screening of, e.g., potential pharmaceutical compounds. My company is collaborating with CHESS and Cornell researchers to develop improved methods for serial synchrotron crystallography.
Probing protein conformational ensembles and energy landscapes
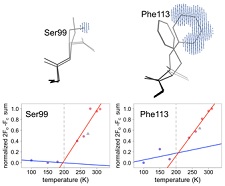
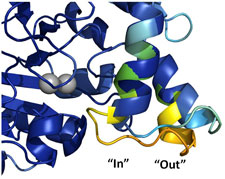
In collaboration with James Fraser's group at UCSF, we have applied our variable temperature crystallography methods to the dynamic enzyme cyclophilin A. Using new tools developed by Lang, Holton, Fraser, and others from Tom Alber's group and by Henry van den Bedem for analyzing electron density maps and performing multiconformer refinement, we have been able to follow the temperature evolution of the conformatoinal ensemble of nearly every amino acid side chain in this enzyme between 300 K and 100 K. The characteristic temperature at which side chain conformers freeze out is highly heterogeneous, ruling out one popular explanation for the protein-solvent glass transition. The temperature dependence of the active site ensemble provides insight into enzymatic mechanism. Variable temperature experiments performed in collaboration with Mike Hough and Richard Strange on the enzyme copper nitrate reductase also provide insight into active site mechanism.
Mapping the Conformational Landscape of a Dynamic Enzyme by XFEL and Multitemperature Crystallography. D. A. Keedy, L. R. Kenner, M. Warkentin, J. B. Hopkins, R. A. Woldeyes, M. C. Thompson, A. S. Brewster, A. H. Van Benschoten, E. L. Baxter, M. Uervirojnangkoorn, J. M. Holton, W. I. Weis, A. T. Brunger, S. M. Soltis, H. Lemke, A. Gonzalez, N. K. Sauter, A. E. Cohen, H. van den Bedem, R. E. Thorne and J. S. Fraser. Elife 4, e07574 (2015).
Active-site protein dynamics and solvent accessibility in native Achromobacter cycloclastes copper nitrite reductase. K. Sen, S. Horrell, D. Kekilli, C. W. Young, T. W. Keal, H. Atakisi, D. W. Moreau, R. E. Thorne, M. A. Hough, and R. W. Strange, IUCrJ 4, 495-505 (2017).
A key issue in interpreting variable temperature crystallographic data is how contacts between protein molecules within the crystal lattice affect temperature-dependent side chain conformations. To investigate this issue, we compared the effects of cryocooling with those of dehydration on crystals of monoclinic lysozyme. We showed that the effects of cooling were broadly similar to the effects of dehydration, for conditions that yielded similar unit cell volume contractions.
Effects of protein crystal hydration and temperature on side chain conformation and heterogeneity. H. Atakisi, D. W. Moreau, and R. E. Thorne, Acta Cryst. D 74, 264-278 (2018).
CryoSAXS for Determining Low-Resolution Protein Structures
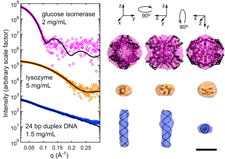
Small-angle X-ray scattering (SAXS) is a widely used technique for obtaining low-resolution structures of proteins, nucleic acids and complexes. SAXS experiments measure molecules in solution at room temperature, without the need for labeling or crystallization. However, radiation damage occurs extremely rapidly, and currently limits the application of SAXS to molecules that can be produced in microgram quantities. As in protein crystallography, cooling SAXS samples to T=100 K might be expected to dramatically reduce radiation damage and sample volume requirements. However, SAXS is exceptionally sensitive to formation of ice and other inhomogeneities.
Building upon our expertise in cryocooling, cryopreservation and aqueous glass formation, we have worked with colleague Lois Pollack to demonstrate the feasibility of performing SAXS on flash-cooled protein and nucleic acid samples. Cryocooled proteins and nucleic acids can withstand doses at least two and up to five orders of magnitude larger than room temperature samples. Accurate T=100 K particle envelope reconstructions can be obtained from sample volumes as small as 15 nL, a factor of 1000 smaller than in current practice. Cryo-SAXS will thus enable structure determination of difficult-to-express proteins and biologically important, highly radiation-sensitive proteins including light-activated switches and metalloenzymes. This project is a collaboration with the group of Prof. Lois Pollack in A&EP.
Breaking the radiation damage limit with cryo-SAXS. S. P. Meisburger, M. Warkentin, H. Chen, J. B. Hopkins, R. E. Gillilan, L. Pollack and R. E. Thorne. Biophys. J. 104, 227-236 (2013)
A microfabricated fixed path length silicon sample holder enables robust background subtraction for cryoSAXS. J. B. Hopkins, A. M. Katz, S. P. Meisburger, M. A. Warkentin, R. E. Thorne, and L. Pollack. J. Appl. Cryst. 48, 227-237 (2015).
Quantifying radiation damage in biomolecular small-angle X-ray scattering. J. B. Hopkins and R. E. Thorne. J. Appl. Cryst. 49, 880-890 (2016).
Temperature-dependent studies of protein structure
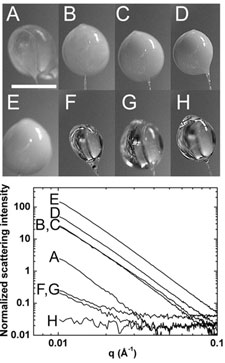
Proteins are dynamic many-degree-of-freedom entities, described by a complex, multi-tiered energy lanscape, and at biological temperature existing in an ensemble of conformations. Despite this fact, 98% of protein structures are determined using cryocooled crystals. In the more than 60 year history of protein crystallography, the structures of only a handful of proteins have been determined at temperatures other than 300 and 100 K. The temperature evolution of structure - especially between T=300 and the protein-solvent glass transition near 200 K - can in principle provide insight into conformational energy landscapes, the stability of protein folds, the structure and mechanism of allosteric networks, the role of water structure and entropy, and possible mechanisms of enzymatic action. The obstacle to such studies has been ice formation, which has prevented structure determination between ~260 and ~180 K.
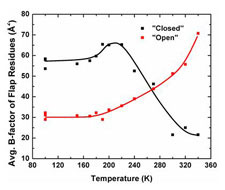
Using insights from our slow cooling studies, we have developed methods for full variable temperature data collection at all temperatures between T=300 K and 100 K. We have used these methods to study structure evolution near the active site in the enzyme urease, and to understand them mechanisms of radiation damage to protein crystals.
Glass transition in thaumatin crystals revealed through temperature dependent radiation sensitivity measurements. M. Warkentin and R. E. Thorne, Acta Cryst. D 66, 1092-1100 (2010).
Dark progression reveals slow timescales for radiation damage between T=180 and 240 K. M. Warkentin, R. Badeau, J. Hopkins and R. E. Thorne, Acta Cryst. D 67, 792-803 (2011).
Spatial distribution of radiation damage to crystalline proteins at 25 to 300 K. M. Warkentin, R. Badeau, J. B. Hopkins and R. E. Thorne, Acta Cryst. D68, 1108-1117 (2012).
Technology and methods for serial synchrotron crystallography
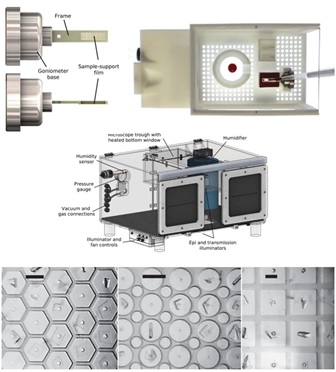
In serial crystallography at X-ray free-electron laser (XFEL) sources, a single frame of diffraction data is collected from each of an enormous number of small crystals, and then these frames are merged to yield a complete data set sufficient for biomolecular structure determination. Methods, hardware, and software have evolved rapidly in the last decade. However, XFEL-based approaches are only suitable for proteins that can be be produced in large quantities and that form suitably monodisperse crystals. Serial crystallography at synchrotron sources can be orders of magnitude more sample efficient, and can use diverse crystals generated in routine crystallization experiments. My company, working with CHESS staff scientists and other Cornell researchers, has been developing improved methods and tools for serial data collection at synchrotrons.
Integrated sample handling and mounting system for fixed-target serial synchrotron crystallography," G. Illava, R. Jayne, A. D. Finke, D. Closs, W. Zeng, S. K. Milano, Q. Huang, I. Kriksunov, P. Sidorenko, F. W. Wise, W. R. Zipfel, B. A. Apker, and R. E. Thorne. Acta Cryst. D 77, 628-644 (2021).