Making Movies of Proteins in Action
Millisecond time-resolved quenching crystallography
Making atomic-resolution movies of biomolecules (e.g., enzymes) in action has been a long-term goal in structural science.
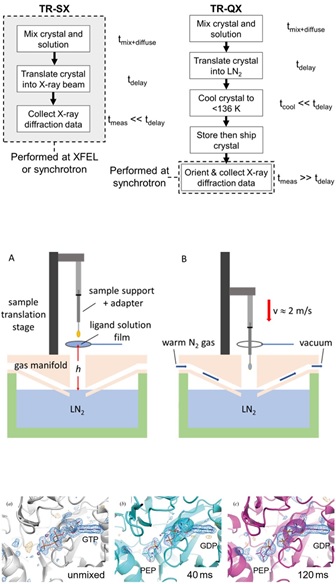
To make a movie of a chemical reaction within a protein crystal, the reaction must be initiated in every molecule within the crystal in a time short compared with that of the reaction, and then a series of snapshots acquired with increasing time delays. In current serial crystallography methods, each "snapshot" records the diffraction pattern from each crystal in only a single, random crystal orientation. A very large number of snapshots from an equally large number of crystals must be acquired to obtain enough coverage of reciprocal space to reconstruct the 3D atomic structure at each time delay. Since the X-rays damage (or in the case of XFELs, destroy) the crystal, data for only one time point can be collected per crystal. As result, protein and crystal consumption (10^4 to 10^7 crystals per time point) is enormous, complex apparatus is required to efficiently deliver those crystals into the X-ray beam, and only a handful of protein systems have been studied in the last decade.
An alternative approach, used in spectroscopic methods and in cryo-EM, is to initiate the reaction and then rapidly cool the sample after a time delay, quenching the reaction and capturing an intermediate state. That state can then be examined at a later time and place. Data collection is not constrained by the reaction timescale, and so can be done in the most efficient way possible, maximizing the amount of useful data per sample. Application of this approach in crystallography achieved time resolutions of several seconds, too slow to allow study of the overwhelming majority of reactions of interest.
Building on our understanding of the physics of cryocooling small samples and using an automated cooling instrument we developed, we have improved this time resolution to 40 ms, which allowed us to observe a previously unobserved intermediate state in the enzyme PEPCK. Moreover, we achieved this time resolution using a single crystal per time point, with data collected remotely on a standard cryocrystallography beamline. We are currently working on methodological improvements to reduce our time resolution toward 1 ms, adequate to observe reactions in a large majority of enzyme systems. Our approach promises to turn time-resolved crystallography into a workhorse tool for studying biomolecules in action.
Millisecond mix-and-quench crystallography (MMQX) enables time-resolved studies of PEPCK with remote data collection," J. A. Clinger, D. W. Moreau, M. J. McLeod, T. Holyoak, and R. E. Thorne. IUCrJ 8, 784-792 (2021).
Making atomic-resolution movies of biomolecules (e.g., enzymes) in action has been a long-term goal in structural science.
To make a movie of a chemical reaction within a protein crystal, the reaction must be initiated in every molecule within the crystal in a time short compared with that of the reaction, and then a series of snapshots acquired with increasing time delays. In current serial crystallography methods, each "snapshot" records the diffraction pattern from each crystal in only a single, random crystal orientation. A very large number of snapshots from an equally large number of crystals must be acquired to obtain enough coverage of reciprocal space to reconstruct the 3D atomic structure at each time delay. Since the X-rays damage (or in the case of XFELs, destroy) the crystal, data for only one time point can be collected per crystal. As result, protein and crystal consumption (10^4 to 10^7 crystals per time point) is enormous, complex apparatus is required to efficiently deliver those crystals into the X-ray beam, and only a handful of protein systems have been studied in the last decade.
An alternative approach, used in spectroscopic methods and in cryo-EM, is to initiate the reaction and then rapidly cool the sample after a time delay, quenching the reaction and capturing an intermediate state. That state can then be examined at a later time and place. Data collection is not constrained by the reaction timescale, and so can be done in the most efficient way possible, maximizing the amount of useful data per sample. Application of this approach in crystallography achieved time resolutions of several seconds, too slow to allow study of the overwhelming majority of reactions of interest.
Building on our understanding of the physics of cryocooling small samples and using an automated cooling instrument we developed, we have improved this time resolution to 40 ms, which allowed us to observe a previously unobserved intermediate state in the enzyme PEPCK. Moreover, we achieved this time resolution using a single crystal per time point, with data collected remotely on a standard cryocrystallography beamline. We are currently working on methodological improvements to reduce our time resolution toward 1 ms, adequate to observe reactions in a large majority of enzyme systems. Our approach promises to turn time-resolved crystallography into a workhorse tool for studying biomolecules in action.
Millisecond mix-and-quench crystallography (MMQX) enables time-resolved studies of PEPCK with remote data collection," J. A. Clinger, D. W. Moreau, M. J. McLeod, T. Holyoak, and R. E. Thorne. IUCrJ 8, 784-792 (2021).