Solvent Nanoconfinement, Ice Formation, & Cryocooling of Protein Crystals
Nearly all protein structures are determined using crystals cooled to T=100 K. However, the cooling process disorders the crystals, and the T=100 K structure may differ in important ways from the biologically active form.
Ice in biomolecular crystallography
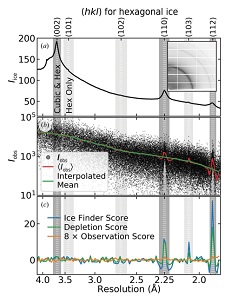
X-ray diffraction from a protein crystal generates spots or peaks at particular angles, and the integrated intensity in each spot gives the amplitude of the corresponding structure factor - related to a Fourier component of the crystal's electron density. When diffraction from ice is also present, the resulting integrated intensities will be biased at the resolutions of the ice rings, and this can lead to errors in molecular modeling. We have improved an algorithm for detecting ice contamination in protein crystal structure factors. We have extended it to allow determination of the type of ice present (hexagonal as frost, near cubic or near hexagonal in surface solvent, stacking disordered inside) and thus diagnose its origin. We then analyzed nearly 100,000 unique entries in the Protein Data Bank (PDB) to determine the prevalence of different types of ice and how this has evolved over the last 20 years. Nearly 16% of PDB entries - generally, based on the best of all X-ray data sets collected in a given study - show evidence of ice biasing in their structure factors.
Ice in biomolecular cryocrystallography. D. W. Moreau, H. Atakisi, and R. E. Thorne. Acta Cryst. D 77, 540-554 (2021).
Solvent nanoconfinement and ice formation in protein crystals
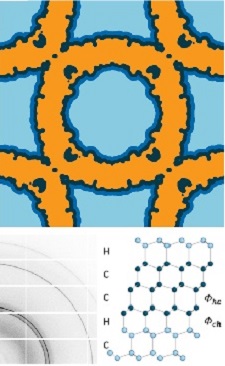
During cooling, ice can form in solvent surrounding a protein crystal, and in solvent residing in the cavities or spaces between proteins within the crystal. By removing the surrounding solvent, we have shown that the freezing temperature and the ice nucleation rate below that temperature are both dramatically suppressed by nanoconfinement within the protein lattice. We have shown that cryoprotectant-free internal solvent (water and a small amount of salt) can be maintainined in a supercooled liquid state down to -100° F (200 K), even in crystals with 7 nm solvent cavities. When internal ice forms, it is neither hexagonal nor cubic ice. Instead, it is stacking disordered with a near 50:50 random stacking of cubic and hexagonal planes, consistent with ice forming under conditions of deep supercooling.
By analyzing diffraction from the protein lattice and from ice within the same crystal, we are able to quantitatively determine the fraction of internal solvent that crystallizes. The fractions determined in three different protein crystal systems indicate that solvent within roughly 0.5 nm of the protein surface never crystallizes.
Ice formation and solvent nanoconfinement in protein crystals. D. W. Moreau, H. Atakisi, and R. E. Thorne. IUCrJ 6, 346-356 (2019).
Solvent flows, conformational changes, and crystal disorder

What happens to the solvent within a protein crystal when it wants to contract less than the protein lattice in whose interstices it resides? We have observed direct manifestations of solvent flows in protein crystals during and after cooling using X-ray crystallography. During rapid cooling, excess solvent is expelled from the unit cells and accumulates in internal crystal regions, decreasing overall crystal order. If the crystal temperature remains above ~200 K, the internal solvent remains liquid and conformational relaxation and repacking of the protein molecules can occur. In the case of apoferritin, this repacking increases the volume of the unit cell, allowing solvent that was pushed out to return, and leading to a recovery of the initial crystal order. These demonstrate that solvent redistribution is a major cause of cooling-induced protein crystal disorder and suggest approaches to reducing it.
Solvent flows, conformation changes and lattice reordering in a cold protein crystal. D. W. Moreau, H. Atakisi, and R. E. Thorne. Acta Cryst D 75, 980-995 (2019).
Character and origin of cooling induced disorder in protein crystals

Flash Cooling and Annealing of Protein Crystals. S. Kriminski, C. L. Caylor, M. C. Nonato, K. D. Finkelstein and R. E. Thorne, Acta Cryst. D58, 459-471 (2002).
Heat transfer from protein crystals
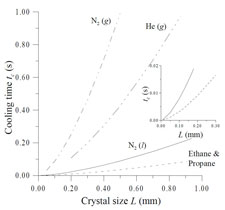
We have analyzed heat transfer from protein crystals during cooling in cold gas streams and during plunging into liquid cryogens, and determined the scaling of cooling rates and internal temperature gradients with several relevant experimental parameters. This analysis allows maximum feasible cooling rates as well as maximum crystal temperature rises due to X-ray absorption to be estimated.
Heat Transfer from Protein Crystals: Implications for Flash Cooling and X-ray Beam Heating. S. Kriminski, M. Kazmierczak and R. E. Thorne, Acta Crystallographica D 59,697-708 (2003).
Ultra-fast and slow cooling
Protein conformation is temperature dependent, and so cooling crystals leads to changes in structure. Protein crystals typically contain between 30 and 90% water. On cooling, this water may crystallize, producing ice diffraction that interferes with that from the protein, and damaging the protein lattice. One might guess that there are two favorable limits for cooling crystals from room temperature to T=100 K:
However, cooling rates achieved in standard practice are in neither of these favorable limits.
Ultra-fast cooling:
The fastest cooling rates are obtained by plunging samples from air into a cryogenic liquid such as nitrogen, propane or ethane. We have demonstrated a simple plunge cooling method that increases cooling rates for small samples by a factor of 20-100 over previous best practice. These cooling rates - 20,000 to 100,000 K/s - allow even protein-free aqueous solutions to be vitrified with very small (<10%) cryoprotectant concentrations.
Above any cold liquid surface there will be a layer of cold gas, formed by conduction and convection and, if the cryogenic liquid is near its boiling point, by evaporation. For sufficiently small samples, most cooling occurs in this gas layer, not in the liquid, at a rate determined by heat transfer to the gas. Our measurements showed that "sufficiently small" is smaller than roughly 500 microns - which is larger than nearly all protein crystals. By simply blowing this cold gas layer away immediately prior to the sample plunge, we increased cooling rates from ~1000 K/s to ~20,000 K/s for ordinary size crystals.
Hyperquenching for Protein Crystallography. M. Warkentin, V. Berejnov, and R. E. Thorne, J. Appl. Cryst. J. Appl. Cryst. 39, 805-811 (2006).
Slow cooling:
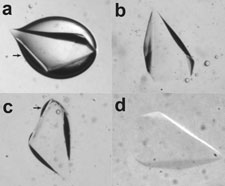
The obvious obstacle to successful slow cooling is ice crystal nucleation and growth. It has long been assumed - and observed - that protein crystals must be rapidly cooled to prevent ice formation. We have shown that protein crystals with solvent contents up to at least 65% can be cooled to T=100 K at only 0.1 K/s without appreciable ice formation, often without any penetrating cryoprotectants. This remarkable result indicates that ice nucleation is strongly suppressed by confinement in the nanoporous network found inside protein crystals, and that nearly all ice seen in diffraction patterns nucleates in the solvent outside the crystal.
Slow cooling of protein crystals. M. Warkentin and R. E. Thorne, J. Appl. Cryst. 42 944-952 (2009).
Density and electron density of aqueous glasses at cryogenic temperatures
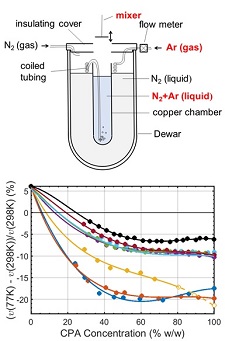
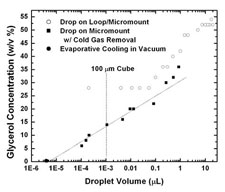
Protein crystals are typically grown or soaked in aqueous solutions containing between 15% and 50% w/v of cryoprotectants such as glycerol or ethylene glycol. These solutions contract by a different amount than the solvent cavities within a protein crystal's unit cell during cooling, and this differential contraction can damage the crystal. We have made the first measurements of the cryogenic-temperature densities of several aqueous cryoprotectant solutions at concentrations below 50% w/w using a novel buoyancy-based method, and used these to determine the volume contraction of these solutions on cooling to cryogenic temperature. We have also determined the room and cryogenic temperature electron densities of these solutions, which determine the strength of diffraction contrast (related to diffraction signal to background) in both crystallography and small-angle X-ray scattering.
Thermal contraction of aqueous glycerol and ethylene glycol solutions for optimized protein crystal cryoprotection. C. Shen, E. F. Julius, T. J. Tyree, D. W. Moreau, H. Atakisi, and R. E. Thorne. Acta Cryst. D 72, 742-752 (2016).
Measuring the densities of aqueous glasses at cryogenic temperatures. C. Shen, E. F. Julius, T. J. Tyree, R. Dan, D. W. Moreau, and R. E. Thorne, JoVE 124, e55761 (2017).
Density and electron density of aqueous cryoprotectant solutions at cryogenic temperatures for optimized cryoprotection and diffraction contrast. T. J. Tyree, R. Dan, and R. E. Thorne. Acta Cryst. D 74, 471-479 (2018).
Cryocrystallography without cryoprotectants

Protein crystallographers have long assumed that penetrating cryoprotectants are needed to prevent ice formation inside their crystals, and much time and effort is expended finding suitable cryoprotectants and concentrations.
We have shown that essentially all crystals can be successfully cooled to T=100 K without any penetrating cryoprotectants using our hyperquenching method, provided that the external solvent is removed, e.g., using oil, blotting or dehydration.
A general method for hyperquenching protein crystals. M. Warkentin and R. E. Thorne, J. Struct. Funct. Gen. 8, 141-144 (2007).
For many protein crystals (with solvent contents up to at least 65%), no penetrating cryoprotectants are necessary even when crystals are cooled very slowly, provided all external solvent is removed.
Slow cooling and temperature-controlled protein crystallography. M. Warkentin and R. E. Thorne, J. Struct. Funct. Gen. 11, 85-89 (2010).
Solvent nanoconfinement, solvent flows, and ice Formation in protein crystals
During cooling, ice can form in solvent surrounding a protein crystal, and in solvent residing in the cavities or spaces between proteins within the crystal. By removing the surrounding solvent, we have shown that the freezing temperature and the ice nucleation rate below that temperature are both dramatically suppressed by nanoconfinement within the protein lattice. We have shown that cryoprotectant-free internal solvent (water and a small amount of salt) can be maintainined in a supercooled liquid state down to -100° F (200 K), even in crystals with 7 nm solvent cavities. When internal ice forms, it is neither hexagonal nor cubic ice. Instead, it is stacking disordered with a near 50:50 random stacking of cubic and hexagonal planes, consistent with ice forming under conditions of deep supercooling.
By analyzing diffraction from the protein lattice and from ice within the same crystal, we are able to quantitatively determine the fraction of internal solvent that crystallizes. The fractions determined in three different protein crystal systems indicate that solvent within roughly 0.5 nm of the protein surface never crystallizes.
Ice formation and solvent nanoconfinement in protein crystals. D. W. Moreau, H. Atakisi, and R. E. Thorne. IUCrJ (2019) in press.
New crystal mounting methods for cryo- and room-temperature crystallography

We developed microfabricated ultra-X-ray transparent tools for crystal harvesting and X-ray data collection, especially for harvesting and data collection from microcrystals. These tools have been patented and commercialized by MiTeGen, LLC, and are now used in small molecule and protein crystallography laboratories around the world.
Microfabricated Mounts for Microcrystal Cryocrystallography. R. E. Thorne, Z. Stum, J. Kmetko and K. O'Neill, J. Appl. Cryst. 36, 1455-1460 (2003).
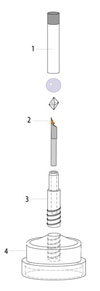
Part of the reason why room-temperature X-ray data collection has all but been abandoned is the difficulty of mounting and handling crystals in convention X-ray capillaries. We have developed a new method for room-temperature sample mounting that allows crystals to be mounted in seconds with essentially 100% chance of success, and to be shipped to the synchrotron for data collection. This mounting method has been patented, and has been commercialized by MiTeGen, LLC.
A new sample mounting technique for room-temperature macromolecular crystallography. Y. Kalinin, J. Kmetko, A. Bartnik, A. Stewart, R. Gillilan, E. Lobkovsky and R. E. Thorne, J. Appl. Cryst. 38, 333-339 (2005).